
La conformité des dispositifs médicaux est essentielle pour garantir que chaque produit est sûr et efficace pour l’usage prévu. Elle englobe divers aspects, notamment réglementaire, clinique et technique, assurant que le dispositif respecte les exigences légales et normatives en vigueur.
Les normes et régulations en vigueur en Europe
Dans un premier temps, le fabricant de dispositifs médicaux doit répondre aux exigences générales du règlement de l’Union Européenne (2017/745). Il existe en effet plusieurs normes qui s’adaptent à chaque type de dispositif médical, mais il y a une liste de normes harmonisées qui sont utilisées le plus souvent par les fabricants. Par exemple, on retrouve la norme EN ISO 13485 qui annonce les exigences liées au système de management de la qualité. Aujourd’hui, les organismes de régulation jouent un rôle très important dans la surveillance et l’approbation des dispositifs avant leur mise sur le marché. Nommés par un Etat membre de l’Union Européenne, ces organismes notifiés ont pour mission de procéder à l’évaluation des dispositifs pour s’assurer de leur sécurité et de leur parfaite conformité aux exigences, pour ensuite délivrer le marquage CE

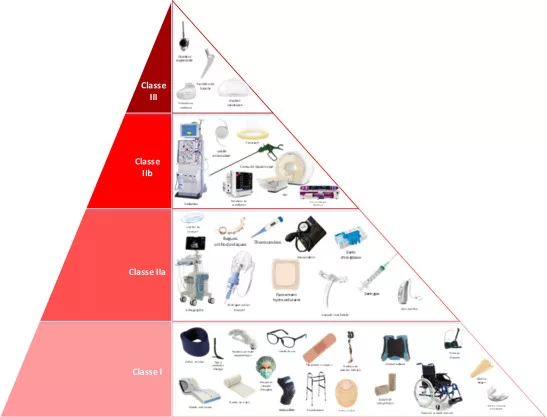
Les différentes classes et niveaux de conformité
Les dispositifs médicaux (DM) sont classés en différentes catégories selon leur complexité, leur usage et le risque pour les patients et utilisateurs. Leur classification suit généralement le cadre défini par l’Union Européenne, qui comprend quatre classes :
- La classe I pour les dispositifs médicaux de faible risque, telles que les lunettes correctrices
- La classe IIa et IIb pour les dispositifs médicaux de risque modéré à élevé, tels que les lentilles (IIa) de contact puis les respirateurs (IIb).
- La classe III pour les dispositifs médicaux de risque élevé, tels que les cœurs artificiels
Concernant la conformité, la classe I est simplifiée de par le faible risque qu’elle comporte et nécessite seulement une auto déclaration et une surveillance basique.
A partir de la classe II, il peut être requis d’effectuer des essais cliniques, de fournir une documentation plus exigeante et d’être évalué par les organismes notifiés.
Enfin, pour la classe III, il est obligatoire de passer par des essais pré-cliniques et cliniques approfondis, avec une documentation particulièrement détaillée et une évaluation plus rigoureuse par les organismes certifiés. Un suivi strict et continu sera effectué après la mise sur le marché.
Ces approches différentes mettent en perspectives les coûts et les délais plus importants supportés par les dispositifs de classe III.

Les étapes clés du processus de conformité d'un cœur artificiel total
Le processus de conformité pour un dispositif médical aussi complexe qu’un cœur artificiel total implique plusieurs étapes rigoureuses pour garantir sa sécurité et son efficacité avant, pendant et après sa mise sur le marché.
La première étape constitue la recherche et développement (R&D), où l’on commence par conceptualiser le dispositif en définissant ses spécifications techniques et fonctionnelles. La conception des prototypes intègre les exigences réglementaires dès le début.
Puis, viennent les études précliniques. Les tests in vitro sont effectués pour évaluer les matériaux utilisés, leur biocompatibilité et leur durabilité. Ces tests sont suivis d’essais in vivo sur animaux pour vérifier la performance et la sécurité du prototype dans des conditions biologiques réelles.
Après la réussite de ces tests, la phase de documentation et de compilation des données commence pour effectuer un dossier complet et détaillé.
Viennent ensuite les essais cliniques (sur l’Homme) qui sont divisés en plusieurs phases : les tests initiaux sur un petit groupe de patients pour évaluer la sécurité de base puis un groupe plus large pour évaluer l’efficacité et ajuster les paramètres techniques et enfin des essais cliniques à grande échelle pour confirmer la sécurité et l’efficacité.
Après la réalisation des essais cliniques, le dossier technique complet est soumis à un organisme notifié pour évaluation. Une fois certifié, le cœur artificiel entre en phase de production à grande échelle, en respectant les normes de qualité, et de distribution.

Aujourd’hui, Procope Medicals entre en phase d’études pré-cliniques avec comme objectif de réaliser ces tests au cours des 24 prochains mois. Cette étape s’amorce avec un plan d’action rigoureux pour répondre aux exigences de la réglementation de son dispositif médical.
Sources :
https://www.snitem.fr/le-dispositif-medical-dm/dm-et-cadre-reglementaire/la-reglementation-des-dispositifs-medicaux/#:~:text=La réglementation qui encadre le,sur le marché des DM.
https://lne-gmed.com/fr/notified-bodies-role#:~:text=Un organisme notifié est un,à disposition des dispositifs médicaux.